Mitochondrien sind jene Strukturen in den Zellen unseres Körpers, welche die meiste Energie erzeugen. Über 90% der zellulären Gesamt-Energie können hierher stammen. Weiterhin sind viele andere zelluläre Mechanismen mit diesen eifrigen Helfern assoziiert. Die Palette reicht vom Fettsäure-, Porphyrin- oder Aminosäurestoffwechsel, über die Regulation des Calciumhaushalts, sowie dem Schutz vor oxidativen Stress bis hin zum programmierten Zelltod (Apoptose).
Mitochondrien im Einsatz! Thanks to @remotehorst23. License cc0. - unrestricted use allowed
In den letzten Jahren erfreut sich die sogenannte Mito-Medizin immer größerer Bedeutung. Die Mito-Medizin geht davon aus, dass Schäden an den Mitochondrien ursächlich für die Leiden vieler Menschen sind.
Das Spektrum potentiell Mitochondrien-assoziierter Erkrankungen ist dabei sehr weitreichend und umfasst neben einer Vielzahl psychischen Erkrankungen (Autismus, Depressionen, Burn-Out, Alzheimer) auch viele andere Leiden wie Krebs, Diabetes oder allgemeine körperliche Schwäche [1-3].
Bisher ist die Mito-Medizin allerdings noch nicht Gegenstand allgemeinmedizinischer Vorgehensweise und wird daher primär nur durch spezialisierte Praxen und Laboratorien abgedeckt.
Wir wollen aus diesem Grunde in zwei Teilen dieses Feld etwas näher ausleuchten.
Im (heutigen) Ersten Teil gehen wir etwas genauer auf Mitochondrien ein, dabei soll primär geklärt werden:
- Was sind Mitochondrien? Wo kommen sie her?
- Wie funktionieren sie? Was leisten sie für uns?
- Und sind diese wirklich so wichtig?
Im Zweiten Teil werden wir uns näher mit den mitochondrialen Dysfunktionen und Mitopathien an sich befassen, hier werden wir vor allem untersuchen:
- Welche mitochondrialen Krankheiten gibt es?
- Wer leidet am meisten darunter?
- Welche experimentellen Modelle gibt es?
- Lässt sich die Beteiligung der Mitochondrien an diesen Leiden wissenschaftliche nachvollziehen?
- Wie würden sich solche Probleme äußern?
- Was kann man als Betroffener tun und ist dies überhaupt nötig/möglich?
Heute aber, wie gesagt, wollen wir zunächst die Mitochondrien an sich behandeln.
Was sind also Mitochondrien und was sollen wir damit?
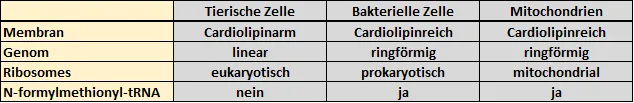
Mitochondrien sind sogenannte Zellorganellen. Organellen sind definitionsgemäß Strukturen in euren Zellen, welche sich durch eigene Membranen von ihrer Umgebung abgrenzen [4]. Durch diese Barriere können Organellen eigene Reaktionsräume ausbilden, um bestimmte Leistungen für die Zelle zu erbringen. Man spricht auch von Kompartimenten (Organellen = Kompartimente). Viele dieser Kompartimente besitzen zumeist eine Doppelmembran, ähnlich der „normalen“ Zellmembran. Die Mitochondrien jedoch verfügen, ähnlich den Chloroplasten in Pflanzen, über zwei Doppelmembransysteme [5]. Außerdem unterscheidet sich die Zusammensetzung der inneren Mitochondrienmembran von dem durchschnittlichen Aufbau tierischer Membransysteme. Die innere Mitochondrienmembran enthält zum Beispiel viel Cardiolipin [6]. Dieser Aufbau ähnelt stark Bakterien [7]! Des Weiteren verfügen die Mitochondrien (genau wie Chloroplasten) über ein eigenes Genom (DNA). Dieses Genom ist wiederum sehr ähnlich dem Genom von Bakterien, denn es ist zum einen ringförmig (das tierische Genom ist bzw. die tierischen Chromosomen sind linear) und enthält sogenannte polycistronische mRNAs [4, 8]. Das bedeutet, dass von Genen Boten-RNAs abgelesen werden, welche gleichzeitig für mehrere Proteine kodieren und auch zusammen abgelesen werden. (Schaut euch in dem Zusammenhang am besten nochmal das Dogma der Molekularbiologie hier an). Bei Tieren gibt es so etwas in der Regel nicht. Außerdem ähneln zum Beispiel tRNAs denen von Bakterien [8]. Es muss aber auch angemerkt werden, dass sich zum Beispiel der Mechanismus der Transkription, der Aufbau der Ribosomen, sowie der Mechanismus der Translation in Mitochondrien von Bakterien und Tieren unterscheidet [2, 9, 10].
Tabelle 1: Tiere, Mitochondrien & Bakterien im Vergleich (Grobe Auswahl!).
Um es kurz zu machen:
Ja lieber Leser, Mitochondrien stammen, trotz gewisser Unterschiede, mit hoher Wahrscheinlichkeit von Bakterien ab, die vor grauer Vorzeit in unsere Zellen eingewandert sind (Endosymbionten-Hypothese, Abb.1) [5].
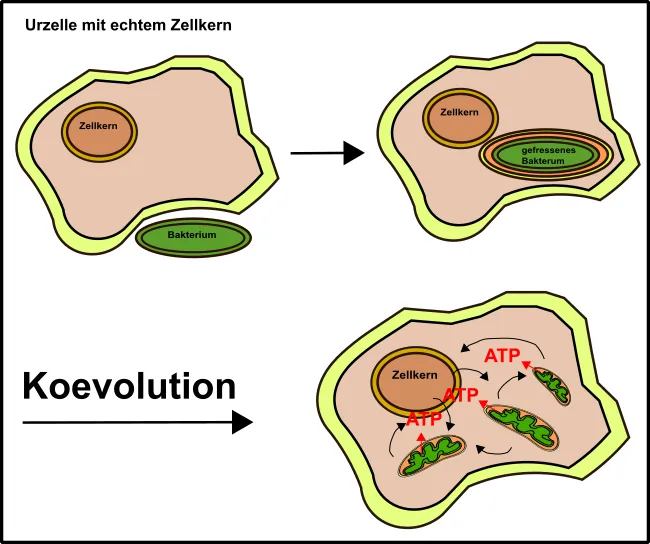
Abb.1: Die Endosymbionten-Hypothese. Erklärung siehe Text. Made by Chapper - unrestricted use allowed
Gleiches gilt übrigens auch für die Chloroplasten. Dabei haben die Bakterien, welche mit unseren Urzellen verschmolzen sind, Teile unserer Membranen mitgenommen und dabei eben zwei Doppelmembransysteme entwickelt (Abb.1).
Der ganze Vorgang muss locker 1,5 Mrd. Jahre her sein [11] und leitet sich allen Anschein nach von α-Proteobakterien ab [10], zu welchen heute Bradyrhizobium japonicum [5] oder auch Vertreter derEssigbakterien [12] zählen. Erstere sind u.a. verantwortlich für die Stickstofffixierung in Sojabohnen (daher so proteinreich). Letztere kennt ihr bestimmt noch aus meinen Kombucha-Artikeln (hier & hier).
Aber warum hat die Urzelle sich das gefallen lassen? Womöglich aus einer Notsituation heraus, denn die Bakterien, die einst mit unseren Vorfahren verschmolzen, brachten besondere Vorteile mit. Besonders hervorzuheben ist dabei ihre Fähigkeit zu Oxidativen Phosphorylierung (OxPhos) [3].
Die Oxidative Phosphorylierung ist ein Mechanismus, welchen ich bereits schon häufiger angesprochen habe (z.B. hier, hier oder auch hier). Es handelt sich um die Übertragung von in der Nahrung gespeicherten Elektronen auf den Elektronenakzeptor Sauerstoff, wodurch „beiläufig“ ATP gebildet wird. Diese Elektronen treiben auf ihrem Weg zum Sauerstoff hin Pumpen an, welche einem Wasserwerk gleich, sehr starke „elektrochemische Kräfte“ hervorbringen [13]. Diese Kräfte werden schließlich benutzt um ein negativ-geladenes Phosphation an ein ebenfalls negativ-geladenes ADP-Molekül „zu pressen“. Dadurch entsteht das sehr reaktionsfreudige ATP, welches das Gros der zellulären Reaktionen erst ablaufen lässt. Bei diesen Reaktionen löst sich das Phosphat wieder vom ADP und ermöglicht durch die freiwerdende Energie, eben jene energetische Schwelle herabzusetzen, welche die Reaktionen ansonsten schwer oder gar nicht möglich macht [13-15]. Bei diesen Reaktionen handelt es sich u.a. um aufbauende Reaktionen (von Proteinen, Fetten etc.) bis hin zu Bewegungen der Zellen bzw. des Körpers (zum Beispiel Muskelkontraktion), Verdopplung der DNA (Replikation), Reparatur der Zelle und ihrer Bestandteile, Signalweiterleitung, sprich alles was die Zelle tun muss um zu überleben. Denn die Zelle muss, wie eigentlich alles, permanent gewartet und instandgehalten werden. Sie strebt unaufhaltsam auf einen thermodynamischen Gleichgewichtszustand (=Tod) zu, welcher nur durch den Einsatz von Energie verhindert werden kann [13].
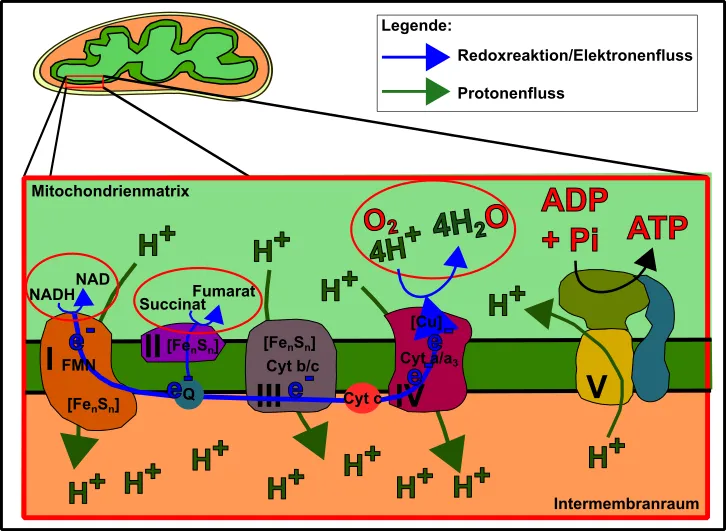
Abb.2 Die mitochondrialeAtmungskette. Made by Chapper - unrestricted use allowed
Nun fragt sich der Leser sicherlich was denn die Urzelle vorher ohne die Mitochondrien gemacht hat. Gute Frage! Die Zelle hat zuvor wahrscheinlich primär Glykolyse betrieben (Abb.3). Glykolyse ist eine relativ ineffiziente Form der ATP-Generierung. Im Wesentlichen ist die Glykolyse eine Aneinanderreihung von enzymatischen Schritten zum Abbau von Zucker, an deren Ende das Produkt Pyruvat, ein paar Reduktionsäquivalente (das sind an einen Carrier gebundene Elektronen), sowie ein wenig ATP steht. Wegen dieser offensichtlichen Kette an Reaktionen, wird dieser Mechanismus auch als Substratketten-Phosphorylierung bezeichnet. Die Elektronen, welche von den Carriern (NAD) abgefangen worden sind, wurden einst womöglich einfach auf Pyruvat oder andere Produkte übertragen und somit „verschwendet“. Dies war allerdings wichtig, damit die Carrier für die nächste Reaktionsfolge wieder zur Verfügung stehen konnten [13, 15-17].
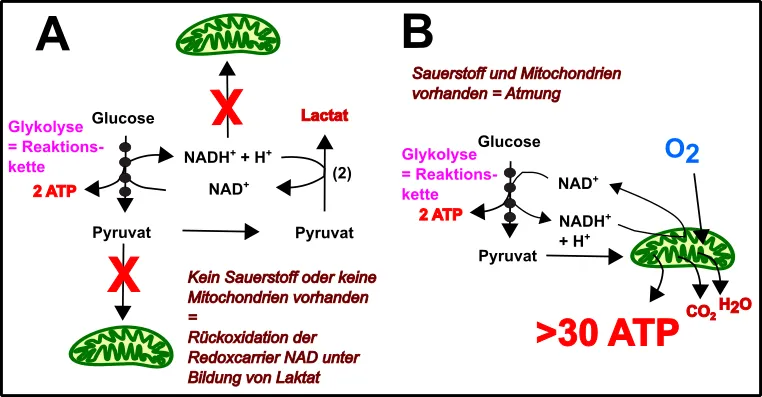
Abb.3 Glykolyse & Mitochondrien: Eine perfekte Energieproduktionseinheit! Made by Chapper – unrestricted use allowed
Seit die Mitochondrien Teil „unserer“ Zellen sind, haben diese die Möglichkeit [13, 16]:
1. die Elektronen mit Hilfe der Carrier in die Atmungskette einzuspeisen, um so noch mehr ATP rauszuholen,
2. konnte nun das Pyruvat weiter zu Kohlenstoffdioxid (CO2) abgebaut werden, um noch mehr Elektronen herauszupressen, was wiederum mehr ATP bedeutet und
3. wurde die Versäuerung der Zelle etwas vermindert, denn nach Übertragung der Elektronen auf Pyruvat entsteht Laktat und dies senkt den pH-Wert.
Darüber hinaus konnten aber noch weitere Vorteile erzielt werden (Abb.4) [15]:
eine effiziente Verwertung von Fettsäuren zur Energiegewinnung war nun problemlos möglich
Aminosäuren (die Bausteine der Proteine) konnten nun effizienter verwertet werden
auch konnten die Aminosäuren über den Stoffwechselschritt des Citratzyklus effizient auf- und umgebaut werden
weitere Stoffwechselwege wie jener der Ketonkörper waren nun verwendbar, dadurch war die Zelle noch flexibler in der Lage auf Änderungen im Nährstoffangebot zu reagieren
die Herstellung von Vitaminen für besondere Stoffwechselwege (wie Vitamin B12) war nun möglich
Entgiftung
uvm.
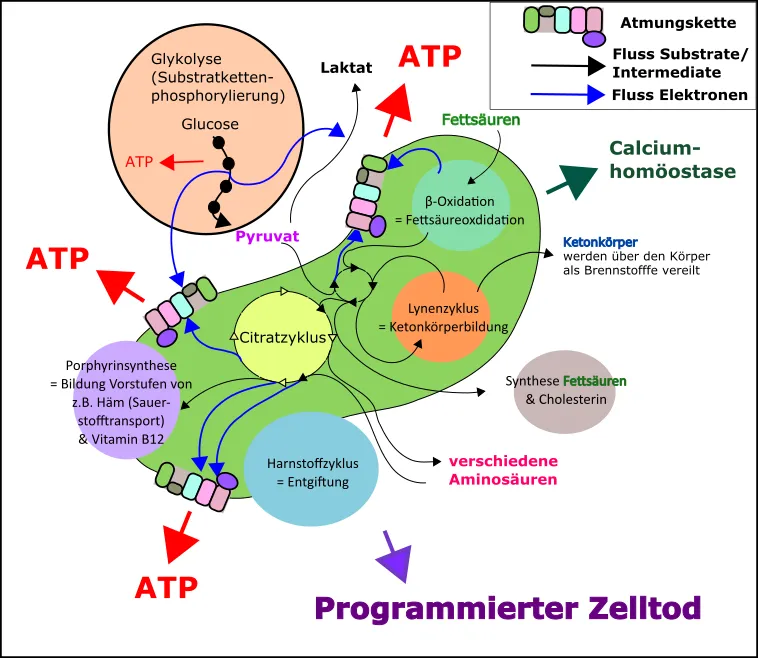
Abb.4 Kleine Auswahl an Interaktionen und Leistungen der Mitochondrien (grüne Struktur in der Mitte des Bildes) für die menschliche Zelle. Beachtet bitte, dass dies nu rein grober Überblick ist und zur besseren “Übersicht” zahlreiche wichtige Details ausgespart wurden. Made by Chapper – unrestricted use allowed
Die Mitochondrien gaben den Urzellen somit ein ungeheuren Innovationsvorteil mit, den vorher nur Bakterien, aber nicht die Zellen mit dem „Echten Zellkern“ die sogenannten Eukaryoten hatten.
Und was bekamen die Mitochondrien im Gegenzug [2, 15, 18]?
sie waren eingebettet in eine weitere Schutzhülle und mussten sich um die Änderungen der äußeren Bedingungen keine großen Sorgen mehr machen, sie waren Teil der zellulären Homöostase (Konstanz des Milieus)
sie wurden auch regelmäßig mit Nahrung versorgt (Versorgung)
außerdem konnten sie große Teile ihrer Genetik in den neuen „Superkern“ auslagern und mussten sich nur noch um ein paar „Specials“ kümmern (Arbeitsteilung/Outsourcing)
Die Mitochondrien konnten somit zahlreiche lästige Aktivitäten gewissermaßen „outsourcen“ und sich voll und ganz auf ihre Kernkompetenzen, allen voran die oxidative Phosphorylierung konzentrieren.
Mit dieser Besonderheit ergaben sich allerdings auch ein paar Probleme, denn der Mechanismus der optimalen Sauerstoffreaktion hin zum Wasser, ist eine idealisierte Wunschvorstellung. Sauerstoff nimmt die Elektronen häufig „unvollständig“ auf, wodurch dieser in äußerst reaktive Zustände gelangt, welche eine sehr hohe Tendenz zur Reaktion mit anderen zellulären Komponenten aufweisen.
Da diese als reaktive Sauerstoffspezies (kurz ROS)(Abb.5) bezeichneten Spezies arge Probleme mit sich bringen, muss die Zelle versuchen [19]:
1. die Übertagung der Elektronen auf den Sauerstoff so effizient wie möglich abzuwickeln und
2. die anfallenden reaktiven Sauerstoffspezies so schnell wie möglich zu entschärfen.
Der erste Fall (Elektronentransport) wird durch die Struktur und Integrität der Atmungskette ermöglicht. Schaut in dem Zusammenhang ruhig nochmal Abb.1 genau an. Die oben angesprochenen Pumpen, welche durch Transfer von Protonen, jene Kräfte erzeugen, die der ATP-Generierung dienen, sind prinzipiell bestens konstruiert und bestmöglich miteinander verbunden/gekoppelt. Zahlreiche Faktoren wie verschiedenen Metallionen (z.B. eingebettet in schwefelhaltige Cluster, FenSn)), Elektronen-Carrier (wie Ubichinon), die in der Membran „umherschwimmen“ oder auch Proteine wie das hoch-konservierte Cytochrom c tun ihr Bestes, um einen maximalen Protonengradienten (was viel ATP gleichkommt) zu erzeugen und gleichzeitig Sauerstoff an Komplex IV so effizient auf Sauerstoff zu übertragen, damit in der Folge nur „sauberes und frisches“ Wasser entsteht (alles zitieren was geht!).
Leider ist dies ein nahezu unerreichbares Ideal, denn viele Faktoren beeinflussen die Effizienz der Atmungskette, z.B.
Schwankungen im Substratangebot: Mal kommen mehr, mal kommen weniger Reduktionsäquivalente an der Atmungskette an. Die Zelle stellt aber immer genau so viele Atmungskettenkomponenten zur Verfügung wie sie auch tatsächlich braucht. Nichts wird verschwendet! Auf rapide Änderungen kann daher oft nicht sofort reagiert werden [16].
Engpässe im Elektronenfluss: Treffen zu viele Elektronen an der Atmungskette ein, dann können sich diese an bestimmten Komplexen „stauen“. Die Folge ist dann eine „Entladung“ zum Beispiel an Komplex I & III. Diese sind aber alles andere als dafür ausgelegt, die Elektronen ordnungsgemäß auf Sauerstoff zu übertragen. die Entstehung von ROS ist unausweichlich [20].
Etwaige Mutationen in den Atmungskomplexen oder in Faktoren, welche an deren Aufbau mitwirken: Effizienz ist alles beim Elektronentransport! Spielt einer der Faktoren nicht immer 100% mit, dann kommt es auch umso häufiger zur ROS-Bildung [1, 2].
Engpässe bei der Versorgung mit Atmungskettenkomponenten: Kann die Zelle die Vielzahl der Mineralstoffe, Vitamine oder auch Proteine und Peptide nicht ausreichend zur Verfügung stellen, so sind Staus bzw. Fehlübertragungen unausweichlich [1-3, 16].
Und es gibt noch zahlreiche weitere Ursachen für solche Probleme. Prinzipiell ist die Zelle im Dilemma zwischen ausreichender Energiegewinnung und Komplexität auf der einen Seite und hohen Kosten zur Korrektur und Verhütung von Schäden auf der anderen Seite.
Genau für diesen Zweck hat die Zelle aber zahlreiche Abwehrmechanismen entwickelt [19]:
SOD: SOD steht für die Superoxiddismutase. Die SOD ist ein Enzym, welches sich um eines der häufigsten, aber auch kurzlebigsten Sauerstoffradikale, dem Superoxidradikal, kümmert und in Wasserstoffperoxid (H2O2) umwandelt (Abb.5A).
Katalase: Obwohl Wasserstoffperoxid weniger reaktiv als das Superoxidradikal ist, ist es immer noch ziemlich reaktionsfreudig und dabei noch wesentlich langlebiger. Auch H2O2 muss daher weg und dies arrangiert die Katalase (Abb.5A).
Nun sind die SOD und die Katalase aber nicht immer sofort zur Stelle, wenn mal irgendwo ein Superoxidradikal oder etwas H2O2 anfällt. Diese Substanzen beginnen jedoch oft bereits nach deren Entstehung mit ihrem zerstörerischen Werk, z.B. durch die Oxidation von Proteinen (Abb.5B) [19].
Bevorzugtes Ziel: Schwefel! Innerhalb der Zelle liegen die Schwefelgruppen von Cystein in Proteinenreduziert vor [21, 22]. Kommt nun beispielsweise H2O2des Weges, so oxidiert dies den Schwefel und baut dadurch Disulfidbrücken in die Proteine ein (Abb.5B). Die Proteine verändern dadurch ihre funktionsfähige Struktur und werden unbrauchbar [21, 23]. Im schlimmsten Falle bilden diese sogar Aggregate, ein Kennzeichen von Erkrankungen wie Alzheimer oder Parkinson [24, 25]. Um dies zu verhüten gibt es allerdings das Glutathion-System (GSH/GSSG), welches diese Entgleisung kompensiert (Abb.5B) [13, 19, 21, 23].
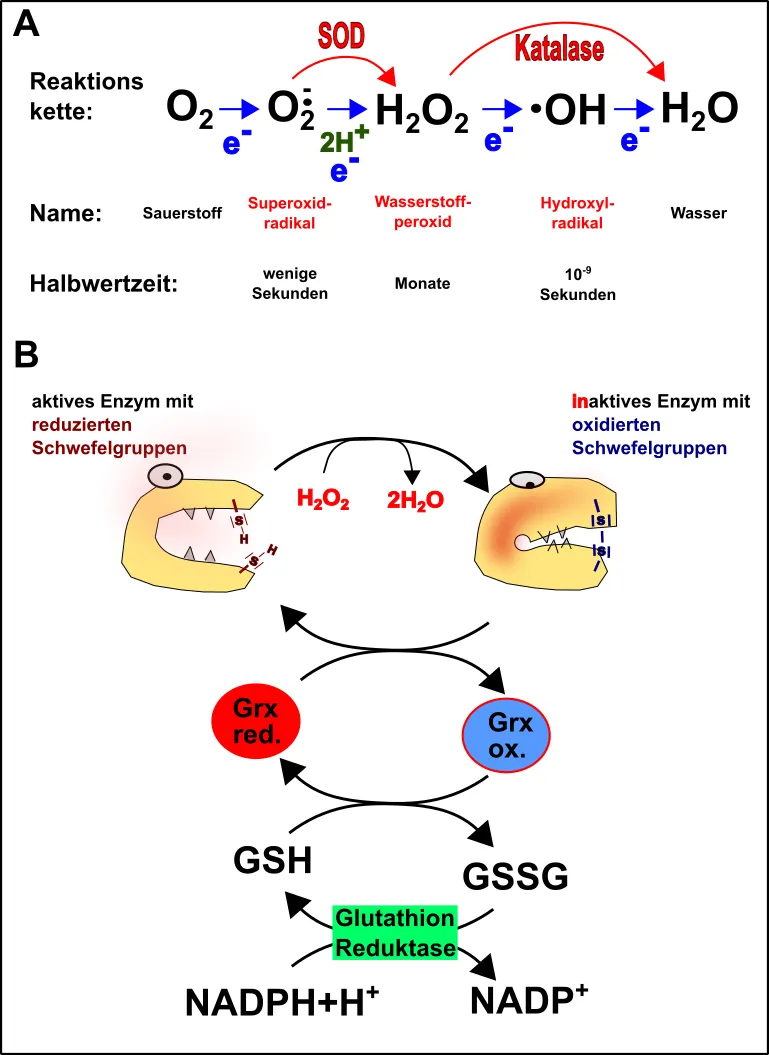
Abb.5 Einführung in die Redoxbiologie. A. Entstehung von reaktiven Sauerstoffspezies (blauer Ast), sowie deren Degradation durch Superoxiddismutase (SOD) und Katalase (roter Ast). B. Inaktivierung von Proteinen durch reaktive Sauerstoffspezies und Rettung durch das Glutathion-System. Grx = Glutaredoxin; GSH = reduziertes Glutathion; GSSG = oxidiertes Glutathion; NADPH (siehe hier). Made by Chapper - unrestricted use allowed
Ein weiteres Tätigkeitsfeld des Glutathion-Systems sind Peroxidradikale. Peroxidradikale entstehen besonders in Membranen, wenn das Superoxidradikal & Co. (vor allem das Hydroxylradikal) zu viel Zeit für ihr finsteres Werk hatten [19]. Bereits nach ihrer Entstehung werden die betreffenden Membranbereiche zerstört. In der Folge reagieren die Peroxidradikale mit allen möglichen Komponenten munter weiter. Außerdem sind Peroxidradikale äußerst stabil und darüber hinaus extrem langlebig und reaktionsfähig. Der totale Alptraum also! Hier kommen Vitamin E (innerhalb der Membran) und Vitamin C (außerhalb der Membran) zum Einsatz. Das anfallende Ascorbinsäureradikal wird seinerseits vom Glutathion-System entschärft. Dieser Prozess, sowie das Recycling des Glutathion-Systems braucht aber reichlich Energie [19].
Ich könnte so jetzt übrigens endlos weitermachen, erinnert euch nur an das 8-Oxogunaosin in der DNA, welches auch oxidiert wird und im schlimmsten Fall zum, vom mir karikierten, Chromosomenmassaker führt (schaut bitte auch hier und hier). Ich möchte es aber erst einmal dabei belassen und euch darauf hinweisen, dass der Preis von viel ATP durch den hohen Preis für Entgiftungs- und Reparaturmechanismen teuer erkauft wird.
Nichtsdestotrotz haben Milliarden von Jahren der Evolution die Rechnung bestanden: Ein flexibleres Nährstoffangebot, ein konstanteres zelluläres Milieu, sowie zahlreiche Zusatzleistungen durch die Mitochondrien, sind einen erhöhten Aufwand bei der zellulären Instandhaltung anscheinend wert.
Apropos Zusatzleistungen: Die Mitochondrien dienen gleichzeitig als “Reset-Knopf“. Hat sich die Zelle bei ihrem Streben nach grenzenloser Macht etwas übernommen, so machen ihr die Mitochondrien im Handumdrehen den Garaus, bevor sie zur Bedrohung für den Rest wird [13, 26, 27]. Dieser Funktion kommen die Mitochondrien übrigens in derart strebsamer Art und Weise nach, so dass ihr davon ausgehen könnt, dass zig Millionen Zellen, just in diesem Moment in eurem Körper das zeitliche segnen ([13]: Allein 1000000000000 Zellen des Blutes pro Tag).
Wir befinden uns jetzt allerdings nicht mehr in der Urzelle, welche munter in ihrer Ursuppe herumschwimmt, sondern bereits in einem vielzelligen Organismus. Hier ist der oben angesprochene Zelltod (Apoptose) ein probates Mittel, nicht nur zur Beseitigung größenwahnsinniger Terrorzellen, sondern unverzichtbares Tool für Entwicklung und Ordnung. Beispielsweise gehen mal locker die Hälfte eurer Neurone in eurer Embryonalentwicklung und darüber hinaus verloren [13]. Dies ist auch wichtig um ein funktionierendes Gehirn überhaupt erst möglich zu machen. Die Mitochondrien wirken somit in einem allumfassenden Kontext bei der Entstehung höherer Lebewesen mit bzw. sind deren Grundvoraussetzung.
Ich denke der Leser versteht eindeutig, dass man die Mitochondrien nicht so einfach auf die leichte Schulter nehmen sollte. Mitochondrien sind neben dem Kern, die heimliche zentrale Einheit der lebenden eukaryotischen Zelle und demnach berechtigtes Diskussionsobjekt, wenn es um das Thema Gesundheit geht.
Und inwieweit diese alten Gefährten tatsächlich einen Einfluss auf unser aller Wohlbefinden haben, das werden wir uns im zweiten Teil genauer anschauen.
Bis dahin
May Da Mitos Be With You.

Euer Chapper
Quellen:
- Wallace, D.C., Mitochondrial DNA mutations in disease and aging. Environ Mol Mutagen, 2010. 51(5): p. 440-50.
- Suomalainen, A. and B.J. Battersby, Mitochondrial diseases: the contribution of organelle stress responses to pathology. Nat Rev Mol Cell Biol, 2018. 19(2): p. 77-92.
- Hill, B.G., et al., Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol Chem, 2012. 393(12): p. 1485-1512.
- W. Janning, E.K., Genetik: Allgemeine Genetik - Molekulare Genetik - Entwicklungsgenetik. 2004: Georg Thieme Verlag.
- Fuchs, Allgemeine Mikrobiologie. Vol. 8. Auflage. 2007: Georg Thieme Verlag.
- Horvath, S.E. and G. Daum, Lipids of mitochondria. Prog Lipid Res, 2013. 52(4): p. 590-614.
- Mileykovskaya, E. and W. Dowhan, Cardiolipin membrane domains in prokaryotes and eukaryotes. Biochim Biophys Acta, 2009. 1788(10): p. 2084-91.
- Taanman, J.W., The mitochondrial genome: structure, transcription, translation and replication. Biochim Biophys Acta, 1999. 1410(2): p. 103-23.
- Koc, E.C. and L.L. Spremulli, Identification of mammalian mitochondrial translational initiation factor 3 and examination of its role in initiation complex formation with natural mRNAs. J Biol Chem, 2002. 277(38): p. 35541-9.
- Greber, B.J. and N. Ban, Structure and Function of the Mitochondrial Ribosome. Annu Rev Biochem, 2016. 85: p. 103-32.
- Embley, T.M. and W. Martin, Eukaryotic evolution, changes and challenges. Nature, 2006. 440(7084): p. 623-30.
- Katsura, K., et al., Asaia siamensis sp. nov., an acetic acid bacterium in the alpha-proteobacteria. Int J Syst Evol Microbiol, 2001. 51(Pt 2): p. 559-63.
- Müller-Esterl, W., Biochemie: Eine Einführung für Mediziner und Naturwissenschaftler. 2004: Spektrum Akademischer Verlag.
- Winter, R. and F. Noll, Methoden der Biophysikalischen Chemie. 1998, Stuttgart: Teubner Studienbücher.
- Peter Karlson, D.D., Jan Koolman, Georg Fuchs, Wolfgang Gerok, Ruth Hammelehle, Karlsons Biochemie und Pathobiochemie. Vol. Auflage: 15. 2005: Thieme.
- Brand, M.D. and D.G. Nicholls, Assessing mitochondrial dysfunction in cells. Biochem J, 2011. 435(2): p. 297-312.
- Püschel, Taschenlehrbuch Biochemie. 2011: Thieme Verlagsgruppe.
- al., G.e., Taschenlehrbuch Physiologie. 2. Auflage ed. 2015: Thieme.
- Kalyanaraman, B., Teaching the basics of redox biology to medical and graduate students: Oxidants, antioxidants and disease mechanisms. Redox Biol, 2013. 1(1): p. 244-57.
- Michalak, K., et al., Treatment of the Fluoroquinolone-Associated Disability: The Pathobiochemical Implications. Oxid Med Cell Longev, 2017. 2017: p. 8023935.
- Brigelius-Flohe, R. and M. Maiorino, Glutathione peroxidases. Biochim Biophys Acta, 2013. 1830(5): p. 3289-303.
- Lopez-Mirabal, H.R. and J.R. Winther, Redox characteristics of the eukaryotic cytosol. Biochim Biophys Acta, 2008. 1783(4): p. 629-40.
- Mari, M., et al., Redox control of liver function in health and disease. Antioxid Redox Signal, 2010. 12(11): p. 1295-331.
- Petrou, M., et al., Amyloid deposition in Parkinson's disease and cognitive impairment: a systematic review. Mov Disord, 2015. 30(7): p. 928-35.
- Wyatt, A.R., et al., Extracellular chaperones and proteostasis. Annu Rev Biochem, 2013. 82: p. 295-322.
- Vanden Berghe, T., et al., Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol, 2014. 15(2): p. 135-47.
- Elmore, S., Apoptosis: a review of programmed cell death. Toxicol Pathol, 2007. 35(4): p. 495-516.
